Neurofibromatosis
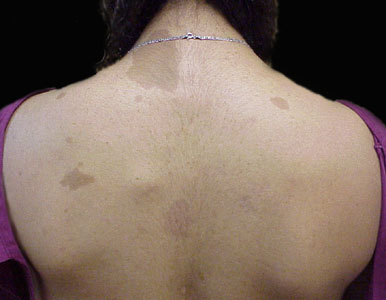
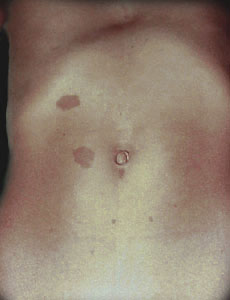
The 'Cafe-au-Lait' (coffe with milk) colored blotches seen on the skin are the most recognized aspect of neurofibromatosis. Even so, these are not only seen in neurofibromatosis. Similar skin findings are found in Oliers's syndrome, Albright's disease, and Jaffe Campanacci syndrome - just to name a few. Also they are common in folks having no syndrome of any kind.
 At one time it was fashionable to use terms like phacodermatoses or neurocutaneous disorder. The gist was a wink wink implication of an embryological fact that is still hard to ignore. In the developing embryo, the nervous system starts out as a stripe
down the center of the top embryologic layer. The embryo has an ectoderm (becomes skin and nervous system) separated from endoderm (guts, lips to tush) by mesoderm (meso means middle, it becomes muscle and bone).
At one time it was fashionable to use terms like phacodermatoses or neurocutaneous disorder. The gist was a wink wink implication of an embryological fact that is still hard to ignore. In the developing embryo, the nervous system starts out as a stripe
down the center of the top embryologic layer. The embryo has an ectoderm (becomes skin and nervous system) separated from endoderm (guts, lips to tush) by mesoderm (meso means middle, it becomes muscle and bone).
The whole thing rolls into a tube so that skin covers all. The
central stripe of neuro-coulda-been-skin sinks in deep and is enveloped. Thus the nervous system is inside not outside. The edge - the very edge - where the neural ectoderm stripe touches plain old skin
ectoderm - that edge is called the neural "crest". 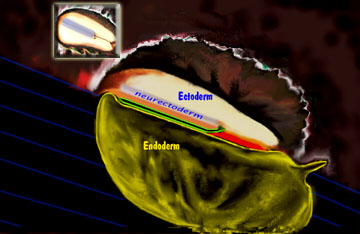 Why crest? Because as the skin and other stuff wells up
to cover the neural stripe, this edge is on that crest. These neural crest cells are restless and do not stay put. They go on to migrate deep and go meandering into the other tissues to head the charge of neural
tissue dispersing throughout other stuff.
Why crest? Because as the skin and other stuff wells up
to cover the neural stripe, this edge is on that crest. These neural crest cells are restless and do not stay put. They go on to migrate deep and go meandering into the other tissues to head the charge of neural
tissue dispersing throughout other stuff.
Neural crest cells have an identifying feature - color. They are the source of pigmented cells. So the common correlation of badly migrated neural paths or cells or whatever and oddball left behind color patches on skin give one pause to consider that the pathology - may vey well include migration defects - regardless of the actual chemistry.
Simple huh? Mmmmmm. The scheme fits many syndromes with odd skin and discoloration seen at birth. Problem is, much of the skin discoloration of neurofibromatosis shows up later - years later. So were these pigmnet cells there all along? Just hiding?
The geneticists have just done a nodding shoulder shrug and go straight to the less understandable geneto -babble (350 kilobase neurofibromin results from a 17q11.2 locus of 60 exons as a GTPase negative regulator of the ras oncogene - but heck, you knew that).
Neurofibromatosis is falls into two main genetic types : Type I or NF1 and type II or NF2.
NF1 is the neurofibromatosis we generally think about when discussing neurofibromatosis - representing 1 case per 4 thousand births (about half as common as club foot). It is the type you see illustrated above, with the skin color patches and with thickened lumpy nerves (neurofibromas), and strangenesses of bones and even organs. Some may even have learning disorders.
NF2 type is way way rare (1/100 as common as NF1). Outright tumors of nerves including those of the ears, eyes, brain, and spinal cord may develop. Hearing loss in a young adult might be the first inkling.
People with freckles. You do not have neurofibromatosis! You are Celtic and driven to dance your feet energetically with your hands stiffly at your sides (or throwing huge logs end over end).
It is the number of large skin discolorations and extent of largeness that typifies the skin findings in neurofibromatosis. Patches on the palms, soles and in the arm pits also have more significance. Despite the gene trail, diagnosis - really meaning confirmation of suspicion in this case - by non-genetic criteria requires certain combinations of clinical findings including number and size of skin discolorations, nerve tumors, bone thinning etc. The confirming criteria may not be met until some time after the condition is clinically suspicious.
The two forms of NF are discrete and don't flip within any given family. They are two discrete dominant gene disorders, not two manifestations of a single disorder. In NF1, half the cases have no parent or family history and are considered to be new mutant forms.
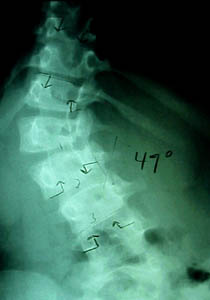 The big significance to pediatric orthopedics is the manifold manifestations, rather than the genetics which are still rather deep and confusing. The penetrance is quite varied. That means some get a whole bunch of skin
markings and have nothing else and some get the nasty something elses with little skin warning. There is a spectrum. Other sorts of tissue tumor tendencies are also known.
The big significance to pediatric orthopedics is the manifold manifestations, rather than the genetics which are still rather deep and confusing. The penetrance is quite varied. That means some get a whole bunch of skin
markings and have nothing else and some get the nasty something elses with little skin warning. There is a spectrum. Other sorts of tissue tumor tendencies are also known.
Our worry in orthopedics is that bones which are involved do not seem to know what they are. They don't heal. You can bone graft defects only to have the defects eat the bone graft pffft - gone. Bones burried in the ground last for millenia. Sharks can't digest bone. Somehow, someway, neurofibromatous tissue occasionally has the cell chemistry that we would only expect of an osteoclast - the ability to quickly digest bone.
So when a neurofibromatous skeletal deformity is seen and treatment planned, the wild card is that the basis of treatment - bone healing - may fail. For that reason, surgical planning often tries to span or work around or away from the involved site - in a quest for properly responsive tissue.
The many manifestations of neurofibromatosis and the depth of known facts can overpower even a totally dedicated site. There are many neurofibromatosis links to be found on the web. Support organizations are out there as well as some deep science web sites. The one from the CDC is quite comprehensive with additional links ( www.cdc.gov/genomics/hugenet).